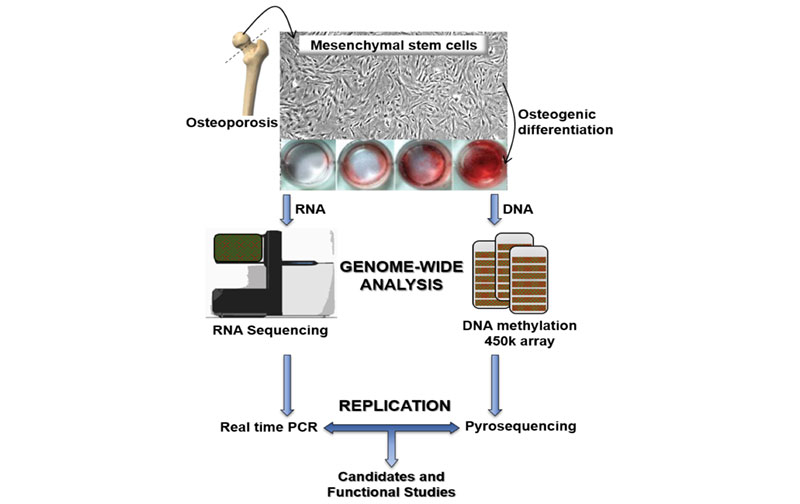
Osteoporosis is the most prevalent bone disorder, characterized by low mineral density, more porous bone and consequently an increased susceptibility to fractures. Specifically, it is caused by an imbalance in the bone remodelling process, with a predominance of osteoclast activity over osteoblastic bone formation. Osteoblasts are responsible for bone formation and this type of cells are the result of the differentiation of Mesenchymal stem cells (MSCs), which are pluripotent stem cells with the capacity to differentiate into a wide variety of cells, including osteoblasts, adipocytes, chondrocytes and myocytes. Thus, MSCs are essential for the maintenance of bone mass. Epigenetic mechanisms are essential for cell differentiation and are also important for the pathogenesis of different diseases. Theoretically, the functional capacity of MSCs may be compromised in elderly people, in relation to the epigenetic changes associated with aging. However, the role of these cells in the pathogenesis of osteoporosis is not well established. Therefore, the aim of this thesis was to analyze DNA methylation marks, gene expression (protein coding and non-protein coding) and the differentiation capacity of bone marrow MSCs of patients with osteoporotic hip fractures.
We obtained MSCs from the femoral heads of women undergoing hip replacement surgery due to hip fractures and from controls with hip osteoarthritis. DNA methylation was explored with the Infinium 450K array (Illumina). Transcriptome analysis was performed by RNA sequencing. DNA methylation values were replicated by pyrosequencing and gene expression values from RNA sequencing were replicated by real-time PCR. In addition, the proliferation rate of MSCs was assessed by the Ki67 positivity and the reduction of MTT. Their ability to differentiate in osteogenic medium was evaluated by staining with alizarin red and quantification of alkaline phosphatase activity.
MSCs of patients with fractures showed greater cell proliferation and gene expression of the master regulator genes RUNX2 and OSX. When cultured in osteogenic medium, MSCs of patients with fractures showed an altered differentiation capacity, with reduced alkaline phosphatase activity and a deficient accumulation of a mineralized matrix. Genome-wide methylation analysis showed that differentially methylated sites, in MSCs from patients with osteoporotic fractures, were enriched in enhancer regions. Moreover, those genes with differential methylated enhancers were overrepresented among those showing differential expression. Gene ontology enrichment analysis revealed that genes with hypomethylated enhancers and upregulated expression in fractures were overrepresented in pathways related to bone. Concerning the long non-protein coding specific gene expression, we found that most transcripts were antisense type. Then we associated the protein coding genes in cis position with those antisense lncRNAs differentially expressed and showed that they were overrepresented in pathways related to bone formation.
Overall, our results suggest that both epigenetic mechanisms, DNA methylation marks of enhancer regions and lncRNAs expression, play an important role in the regulation gene expression pattern of MSCs derived from patients with osteoporosis. These pathways will enhance our understanding of the pathogenesis of osteoporosis, and will also help to characterize new candidates for an anabolic bone therapy.
